Construyendo un modelo deterministico simple
Última actualización: 2026-04-28 | Mejora esta página
Hoja de ruta
Preguntas
- ¿Cómo construir un modelo simplificado de zika?
Objetivos
Al final de este taller usted podrá:
- Reconocer cómo se construye un modelo determinístico simple mediante ecuaciones diferenciales ordinarias.
- Identificar parámetros relevantes para modelar epidemias de ETV.
- Diagramar la interacción entre los diferentes compartimentos del sistema mediante los parámetros.
- Traducir ecuaciones matemáticas del modelo determinístico a código de lenguaje R.
- Explorar el uso de las simulaciones del modelo para proyectar escenarios de transmisión y potencial impacto de las intervenciones
Pre requisito
Esta unidad tiene como prerequisitos:
- Introducción a R y RStudio
- Introducción a la teoría epidémica
- Historia de las epidemias y las pandemias
- Visualización de datos en R con ggplot
Tabla de contenido
|
Introducción
En esta unidad abordaremos la construcción de un modelo determinístico simple, específicamente para el virus del Zika, una enfermedad que desencadenó una gran epidemia en Latinoamérica y el Caribe, y que fue declarada como emergencia de salud pública de importancia internacional. Utilizando los conocimientos previos de teoría epidémica, construiremos un modelo determinístico tipo SIR que incorpora aspectos demográficos.
Para la construcción de este modelo, aprenderemos sobre la dinámica de interacción entre humanos y vectores, así como de los parámetros fundamentales que rigen estos procesos biológicos. Mediante la construcción de un diagrama, examinaremos estas relaciones y formularemos ecuaciones que describen el comportamiento del sistema. Estas ecuaciones serán la base para simular el modelo en el lenguaje de programación R. A su vez propondremos y modelaremos estrategias de intervención.
Mediante el análisis del modelo, evaluaremos el potencial impacto de esta epidemia en la sociedad, contextualizando algunas de estas intervenciones en América Latina. Además, reforzaremos y aplicaremos temas clave como son: Modelo SIR, Inmunidad de rebaño, Parámetros e intervenciones de control (fumigación, mosquiteros y vacunación) para una Enfermedad transmitida por vectores (ETV).
Tema 6: Modelo Zika en R
En esta sección pondremos en uso el conocimiento adquirido sobre el Zika, los mecanismos involucrados en la transmisión y las ecuaciones del modelo. El objetivo es construirlo en R.
El único paquete que se requiere para el modelamiento es deSolve, el cual permite resolver las ecuaciones diferenciales. Adicionalmente para manejar los datos y graficar los resultados recomendamos usar tidyverse y cowplot.
6.1 Inicio práctica en R
Para iniciar nuestra práctica en R, por favor abra un proyecto de R y cree un nuevo documento. En este documento debemos cargar las funciones que acabamos de explicar. Si tiene dificultades con este proceso por favor repase la unidad Introducción a R.
install.packages(deSolve) # Paquete deSolve para resolver las ecuaciones diferenciales
Una vez instalado el paquete deSolve por favor cárgue los paquetes con las siguientes líneas de código, cópielas en su script de R y ejecútelas.
R
library(deSolve)
library(tidyverse)
library(cowplot)

Recordemos que para crear un modelo necesitamos compartimentos, condiciones iniciales, parámetros y ecuaciones.
Para este modelo en R empezaremos por definir los parámetros, es decir, todos aquellos valores que a través de la investigación se han recopilado y hacen parte del comportamiento de la enfermedad. En la sección anterior hablamos sobre ellos y los completamos en una tabla. Es hora de ingresarlos en R.
Instrucción: Por favor, tome la tabla que trabajó anteriormente e ingrese el valor de cada uno de estos parámetros.
NOTA:
Es importante recordar que en R, puede utilizar objetos previamente creados para realizar cálculos. Por ejemplo, el parámetro muv es el inverso del parámetro Lv, es decir, muv = 1/Lv. Por lo tanto, en R se puede asignar este valor directamente con muv <- 1/Lv. No es necesario realizar la división y asignar el resultado manualmente.
Desafío 1
Instrucción: Por favor, tome la tabla que trabajó anteriormente e ingrese el valor de cada uno de estos parámetros.
R
Lv <- # Esperanza de vida de los mosquitos (en días)
Lh <- # Esperanza de vida de los humanos (en días)
PIh <- # Periodo infeccioso en humanos (en días)
PIv <- # Periodo infeccioso en vectores (en días)
PEI <- # Período extrínseco de incubación en mosquitos adultos (en días)
muv <- # Tasa per cápita de mortalidad de la población de mosquitos (1/Lv)
muh <- # Tasa per cápita de mortalidad de la población de humanos (1/Lh)
alphav <- # Tasa per cápita de natalidad de la población de mosquitos. Por ahora asumiremos que es la misma de tasa de mortalidad.
alphah <- # Tasa per cápita de natalidad de la población de humanos. Por ahora asumiremos que es la misma de tasa de mortalidad
gamma <- # Tasa de recuperación en humanos (1/PIh)
delta <- # Tasa extrínseca de incubación (1/PEI)
Nh <- # Número de humanos. Para este ejercicio proponemos 100.000 humanos. Puede cambiarlos si desea de acuerdo a su contexto.
m <- # Densidad de mosquitos hembra por humano
Nv <- # Número de mosquitos (m * Nh)
R0 <- # Número reproductivo básico
ph <- # Probabilidad de transmisión de un mosquito infeccioso a un humano susceptible después de una picadura.
pv <- # Probabilidad de transmisión de un humano infeccioso a un mosquito susceptible después de una picadura.
b <- sqrt((R0 * muv*(muv+delta) * (muh+gamma)) /
(m * ph * pv * delta)) # Tasa de picadura
betah <- # Coeficiente de transmisión de un mosquito infeccioso a un humano susceptible después de una picadura (ph*b)
betav <- # Coeficiente de transmisión de un humano infeccioso a un mosquito susceptible después de una picadura (pv*b)
TIME <- 1 # Número de años que se va a simular. Para este ejercicio iniciaremos con el primer año de la epidemia.
6.2 Ecuaciones del modelo
Ahora que ya ingresamos al script los parámetros es hora de emplear las ecuaciones que se escribieron antes, las cuales permiten conocer el número de individuos en cada uno de los seis compartimentos en función del tiempo. Tres compartimentos para los humanos y tres compartimentos para los mosquitos, los cuales están identificados por una h (para humanos) y una v (para mosquitos). Para los humanos tenemos los compartimentos; susceptibles, infectados y recuperados (de ahí la palabra SIR) y para los mosquitos los compartimientos son: susceptibles, expuestos e infecciosos (SEI).
Compartimentos
\(S_h\) : Humanos susceptibles
\(I_h\) : Humanos infecciosos
\(R_h\) : Humanos recuperados de la infección (inmunizados frente a nueva infección)
\(S_v\) : Vectores susceptibles
\(E_v\) : Vectores expuestos
\(I_v\) : Vectores infecciosos
Para este modelo emplearemos las siguientes ecuaciones diferenciales:
6.3 Fórmula para calcular \(R_0\) (Número reproductivo básico)
Fórmula necesaria para estimar \(R_0\):
\[ R_0 = \frac{mb^2 p_h p_v \delta}{\mu_v (\mu_v+\delta)(\mu_h+\gamma)} \]
Desafío
Instrucción: Traduzca las ecuaciones a R
Una vez sepamos traducir las ecuaciones a código, se procederá a ejecutar el modelo. Para esto se usará la función ode del paquete deSolve.
Se empezará por crear la función (que luego se usará en el argumento
fun). Para ello es necesario traducir las ecuaciones
del modelo de Zika a R. Abajo encontrará la función ya
construida modelo_zika para que usted reemplace las
ecuaciones que ya completó arriba.
Desafío 3
Instrucción: Reemplace las ecuaciones incompletas del siguiente código por las ecuaciones completas del modelo Zika que trabajó en la instrucción anterior.
R
# Modelo determinístico simple (fun)
modelo_zika <- function(tiempo, variable_estado, parametros) {
with(as.list(c(variable_estado, parametros))
, # entorno local para evaluar derivados
{
# Humanos
dSh <- ____ * Nh - ____ * (Iv/Nh) * Sh - ____ * Sh
dIh <- ____ * (Iv/Nh) * Sh - (____ + ____) * Ih
dRh <- ____ * Ih - ____ * Rh
# Mosquitos
dSv <- alphav * Nv - ____ * (Ih/Nh) * Sv - ____ * Sv
dEv <- ____ * (Ih/Nh) * Sv - (____ + ____)* Ev
dIv <- ____ * Ev - ____ * Iv
list(c(dSh, dIh, dRh, dSv, dEv, dIv))
}
)
}
6.4 Resolviendo el Sistema
Para resolver el sistema es necesario crear los tres argumentos
faltantes (times, parms y
y) para usar la función ode.
Desafío 4
Instrucción: Para times y
parms, copie el código que se encuentra a continuación y
ejecútelo.
R
# Secuencia temporal (times)
tiempo <- seq(1, 365 * TIME , by = 1)
# Los parámetros (parms)
params <- c(
muv = muv,
muh = muh,
alphav = alphav,
alphah = alphah,
gamma = gamma,
delta = delta,
betav = betav,
betah = betah,
Nh = Nh,
Nv = Nv
)
En el código que ejecutó se creó tiempo (times) y parametros (params). Aún nos falta crear el argumento y, el cual desarrollaremos en la siguiente sección.
6.4.1. Condiciones iniciales del sistema (y)
Para definir las condiciones iniciales, recuerde que el escenario a modelar en este ejercicio es para una fecha antes del reporte del primer caso. Por lo tanto estos valores deben reflejar ese contexto.
Discusión
Reflexión: ¿Qué condiciones iniciales tendrían cada uno de los compartimientos?
Desafío 5
Instrucción: Complete los espacios según lo aprendido en el tutorial.
6.4.2 Función ode
Una vez creados todos los argumentos necesarios, es hora de ingresarlos a ode. Recordemos los cuatro argumentos de ode y a que corresponden a:
y:inicio. Vector creado con las condiciones iniciales de los seis compartimentos.
times:tiempo. Vector con la secuencia temporal
fun:modelo_zika. Función que contiene las ecuaciones necesarias para simular el modelo.
parms:parametros. Vector en el cual se recopilaron los parámetros necesarios para simular el modelo.
Desafío 6
Instrucción: Complete los espacios en blanco según lo trabajado hasta el momento.
6.4.3 Introduciendo el primer caso
Ahora que tenemos todos los compartimentos definidos, es hora de ingresar al modelo un individuo infeccioso para iniciar la epidemia.
Discusión
Reflexión: ¿Qué cree que es más probable, que a una población (en otro país) ingrese un humano infeccioso o un mosquito infeccioso?
Para nuestro caso hipotético, vamos a suponer que una persona se infectó en Brasil mientras estaba de turismo y regresó posteriormente a la ciudad ______________ (la ciudad que usted definió al comienzo del ejercicio) siendo el primer sujeto infeccioso en esta población. En este contexto, el compartimento de humanos infecciosos tendrá entonces un individuo, Ih = 1 y el compartimento de humanos susceptibles tendrá un individuo menos, Sh = Nh - 1.
Desafío
Pista: Cambie en R las condiciones iniciales (inicio) de forma que Ih = 1 y Sh = Nh - 1.
6.5 ¡Ahora ejecutaremos el modelo!
Hasta este punto, usted ha completado toda la información faltante en el script para poder ejecutar el modelo.
Aviso
Instrucción: Ejecute cada conjunto de las líneas del script vistas anteriormente, es decir, ejecute las secciones: Lista de parámetros, la sección Modelo determinístico simple (donde construyó el modelo), las secciones Secuencia temporal (tiempo (times)), Los parámetros (parametros (parms)), la sección Condiciones iniciales del sistema (inicio (y)) y la sección final Resuelva las ecuaciones.
Instrucción: Verifique que no aparezca ningún error. En caso de error por favor verifique la escritura del código y que no haya quedado en el código otros caracteres que no corresponden como por ejemplo “_____” los guiones de los espacios para completar.
6.6 Visualizando los resultados
En nuestro curso usaremos ggplot para la visualización de datos. Es importante que repase la Unidad 4. Visualización de datos en ggplot
Hay que recordar que la unidad de tiempo del modelo de Zika está ya definida desde los parámetros como días.
Sin embargo, si usted quisiera visualizar los resultados en semanas,
meses o años puede hacerlo a partir de los resultados del modelo
(salida$time). Para hacerlo, puede usar el siguiente
código.
Desafío 7
Para tener una visualización más significativa de los resultados, convierta las unidades de tiempo de días a años y a semanas.
R
# Cree las opciones de tiempo para años y semanas
salida$años <- salida$time/365
salida$semanas <- salida$time/7
6.7 Visualice y analice la primer epidemia
Empecemos realizando una visualización de la primera epidemia. Dado que es un periodo de un año visualicemos las gráficas en semanas.
Instrucción: Ejecute el código a continuación y analice las gráficas resultantes.
R
# Revise la primera epidemia
p1e <- ggplot(data = salida, aes(y = Ih, x = semanas)) +
geom_line(color = 'firebrick', linewidth = 1) +
ggtitle('Población de humanos infecciosos') +
theme_bw() + ylab('Número') + xlab('Semanas') +
coord_cartesian(ylim = c(0,10000)) #creamos gráfico de población humana infecciosa
p2e <- ggplot(data = salida, aes(y = Rh, x = semanas)) +
geom_line(color = 'olivedrab', linewidth = 1) +
ggtitle('Población humana recuperada') +
theme_bw() + ylab('Número') + xlab('Semanas') +
coord_cartesian(ylim = c(0,100000)) #creamos gráfico de población humana recuperada
plot_grid(p1e, p2e) #creamos gráfico comparativo de la gráfica de población humana infecciosa y población humana recuperada
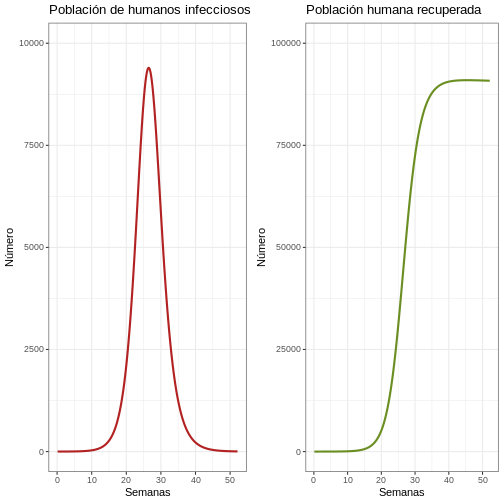
Discusión
Reflexión: ¿Qué puede observar en la gráfica? Observe bien el eje Y. ¿Qué proporción de humanos son infecciosos al mismo tiempo?
Para tener mayor claridad de esto podemos crear gráficas de las proporciones:
R
# Revise la primera epidemia con proporciones
p1p <- ggplot(data = salida, aes(y = Ih/(Sh+Ih+Rh), x = semanas)) +
geom_line(color = 'firebrick', linewidth = 1) +
ggtitle('Población de humanos infecciosos') +
theme_bw() + ylab('Proporción') + xlab('Semanas') +
coord_cartesian(ylim = c(0,1)) #creamos gráfico de población humana infecciosa
p2p <- ggplot(data = salida, aes(y = Rh/(Sh+Ih+Rh), x = semanas)) +
geom_line(color = 'olivedrab', linewidth = 1) +
ggtitle('Población humana recuperada') +
theme_bw() + ylab('Proporción') + xlab('Semanas') +
coord_cartesian(ylim = c(0,1)) #creamos gráfico de población humana recuperada
plot_grid(p1p, p2p) #creamos gráfico comparativo de la gráfica de población humana infecciosa y población humana recuperada
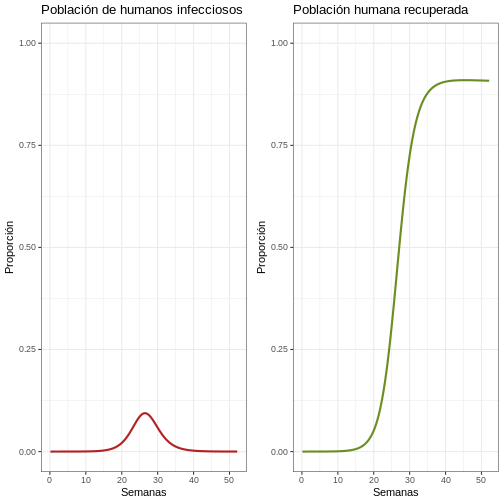
Comportamiento general (Población humana)
Ya observamos la primera epidemia es momento de proyectar la epidemia a un tiempo superior.
Instrucción: Regrese a los parámetros y modifique el parámetro TIME a 100 años. Ejecute el siguiente bloque de código y observe cuántos brotes se producen en la población humana y el tamaño de cada brote.
R
# # Revise el comportamiento general del modelo para 100 años
p1h <- ggplot(data = salida, aes(y = (Rh + Ih + Sh), x = años)) +
geom_line(color = 'grey68', linewidth = 1) +
ggtitle('Población humana total') +
theme_bw() + ylab('Número') + xlab('Años')
p2h <- ggplot(data = salida, aes(y = Sh, x = años)) +
geom_line(color = 'royalblue', linewidth = 1) +
ggtitle('Población humana susceptible') +
theme_bw() + ylab('Número') + xlab('Años')
p3h <- ggplot(data = salida, aes(y = Ih, x = años)) +
geom_line(color = 'firebrick', linewidth = 1) +
ggtitle('Población humana infecciosa') +
theme_bw() + ylab('Número') + xlab('Años')
p4h <- ggplot(data = salida, aes(y = Rh, x = años)) +
geom_line(color = 'olivedrab', linewidth = 1) +
ggtitle('Población humana recuperada') +
theme_bw() + ylab('Número') + xlab('Años')
plot_grid(p1h, p2h, p3h, p4h, ncol = 2)
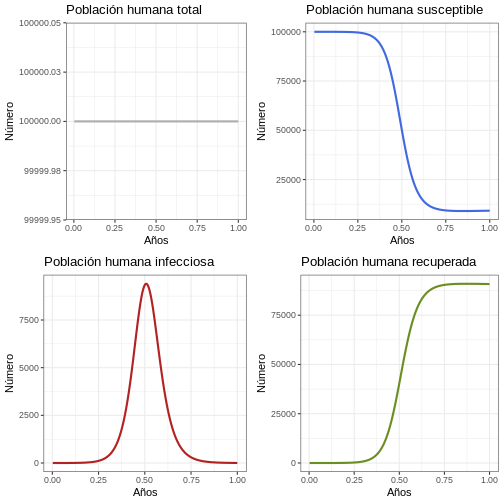
Comportamiento General (Población de mosquitos)
Instrucción: Ejecute el siguiente bloque de código y observe cuántos brotes se producen en la población de mosquitos y el tamaño de cada brote. Compare las gráficas con las gráficas de la población humana.
R
# Revise el comportamiento general del modelo
p1v <- ggplot(data = salida, aes(y = (Sv + Ev + Iv), x = años)) +
geom_line(color = 'grey68', linewidth = 1) +
ggtitle('Población total de mosquitos') +
theme_bw() + ylab('Número') + xlab('Años')
p2v <- ggplot(data = salida, aes(y = Sv, x = años)) +
geom_line(color = 'royalblue', linewidth = 1) +
ggtitle('Población susceptible de mosquitos') +
theme_bw() + ylab('Número') + xlab('Años')
p3v <- ggplot(data = salida, aes(y = Ev, x = años)) +
geom_line(color = 'orchid', linewidth = 1) +
ggtitle('Población expuesta de mosquitos') +
theme_bw() + ylab('Número') + xlab('Años')
p4v <- ggplot(data = salida, aes(y = Iv, x = años)) +
geom_line(color = 'firebrick', linewidth = 1) +
ggtitle('Población infecciosa de mosquitos') +
theme_bw() + ylab('Número') + xlab('Años')
plot_grid(p1v, p2v, p3v, p4v, ncol = 2)
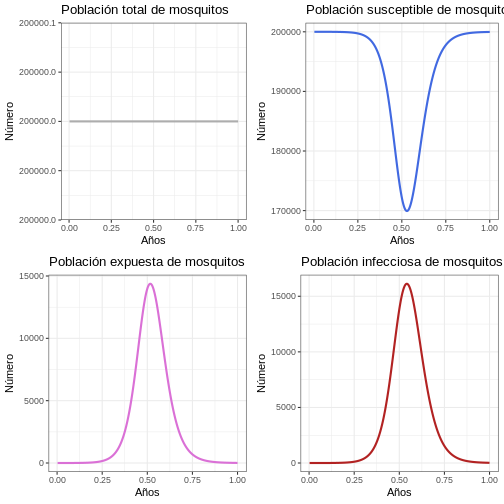
Proporción
Instrucción: Ejecute el siguiente bloque de código y compárelo con las gráficas generadas para la población humana.
R
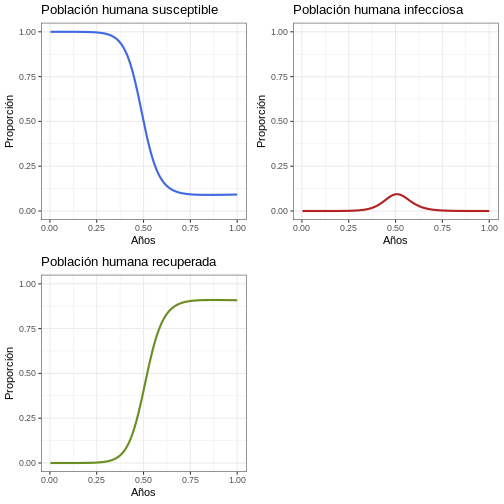
p1 <- ggplot(data = salida, aes(y = Sh/(Sh+Ih+Rh), x = años)) +
geom_line(color = 'royalblue', linewidth = 1) +
ggtitle('Población humana susceptible') +
theme_bw() + ylab('Proporción') + xlab('Años') +
coord_cartesian(ylim = c(0,1))
p2 <- ggplot(data = salida, aes(y = Ih/(Sh+Ih+Rh), x = años)) +
geom_line(color = 'firebrick', linewidth = 1) +
ggtitle('Población humana infecciosa') +
theme_bw() + ylab('Proporción') + xlab('Años') +
coord_cartesian(ylim = c(0,1))
p3 <- ggplot(data = salida, aes(y = Rh/(Sh+Ih+Rh), x = años)) +
geom_line(color = 'olivedrab', linewidth = 1) +
ggtitle('Población humana recuperada') +
theme_bw() + ylab('Proporción') + xlab('Años') +
coord_cartesian(ylim = c(0,1))
plot_grid(p1, p2, p3, ncol = 2)
Puntos Clave
Revise si al final de esta lección adquirió estas competencias:
- Aplicar conceptos como parámetros, \(R_0\) e inmunidad de rebaño, aprendidos en la sesión A del taller
- Traducir fórmulas matemáticas de las interacciones entre los parámetros del modelo a código de R
- Realizar un modelo simple en R para una enfermedad transmitida por vector
- Discutir cambios en las proyecciones del modelo cuando se instauran diferentes estrategias de control de la infección
Contribuciones
- Zulma Cucunuba & Pierre Nouvellet: Versión inicial
- Kelly Charinga & Zhian N. Kamvar: Edición
- José M. Velasco-España: Traducción de Inglés a Español y Edición
- Andree Valle-Campos: Ediciones menores
Referencias
de Carvalho, S. S., Rodovalho, C. M., Gaviraghi, A., Mota, M. B. S., Jablonka, W., Rocha-Santos, C., Nunes, R. D., Sá-Guimarães, T. da E., Oliveira, D. S., Melo, A. C. A., Moreira, M. F., Fampa, P., Oliveira, M. F., da Silva-Neto, M. A. C., Mesquita, R. D., & Atella, G. C. (2021). Aedes aegypti post-emergence transcriptome: Unveiling the molecular basis for the hematophagic and gonotrophic capacitation. PLoS Neglected Tropical Diseases, 15(1), 1–32. https://doi.org/10.1371/journal.pntd.0008915
Chang, C., Ortiz, K., Ansari, A., & Gershwin, M. E. (2016). The Zika outbreak of the 21st century. Journal of Autoimmunity, 68, 1–13. https://doi.org/10.1016/j.jaut.2016.02.006
Cori, A., Ferguson, N. M., Fraser, C., & Cauchemez, S. (2013). A new framework and software to estimate time-varying reproduction numbers during epidemics. American Journal of Epidemiology, 178(9), 1505–1512. https://doi.org/10.1093/aje/kwt133
Duffy, M. R., Chen, T.-H., Hancock, W. T., Powers, A. M., Kool, J. L., Lanciotti, R. S., Pretrick, M., Marfel, M., Holzbauer, S., Dubray, C., Guillaumot, L., Griggs, A., Bel, M., Lambert, A. J., Laven, J., Kosoy, O., Panella, A., Biggerstaff, B. J., Fischer, M., & Hayes, E. B. (2009). Zika Virus Outbreak on Yap Island, Federated States of Micronesia. New England Journal of Medicine, 360(24), 2536–2543. https://doi.org/10.1056/nejmoa0805715
Ferguson, N. M., Cucunubá, Z. M., Dorigatti, I., Nedjati-Gilani, G. L., Donnelly, C. A., Basáñez, M. G., Nouvellet, P., & Lessler, J. (2016). Countering the Zika epidemic in Latin America. Science, 353(6297). https://doi.org/10.1126/science.aag0219
Heesterbeek, J. A. P. (2002). A brief history of R0 and a recipe for its calculation. Acta Biotheoretica, 50(3). https://doi.org/10.1023/A:1016599411804
Lee, E. K., Liu, Y., & Pietz, F. H. (2016). A Compartmental Model for Zika Virus with Dynamic Human and Vector Populations. AMIA … Annual Symposium Proceedings. AMIA Symposium, 2016, 743–752.
Pettersson, J. H. O., Eldholm, V., Seligman, S. J., Lundkvist, Å., Falconar, A. K., Gaunt, M. W., Musso, D., Nougairède, A., Charrel, R., Gould, E. A., & de Lamballerie, X. (2016). How did zika virus emerge in the Pacific Islands and Latin America? MBio, 7(5). https://doi.org/10.1128/mBio.01239-16