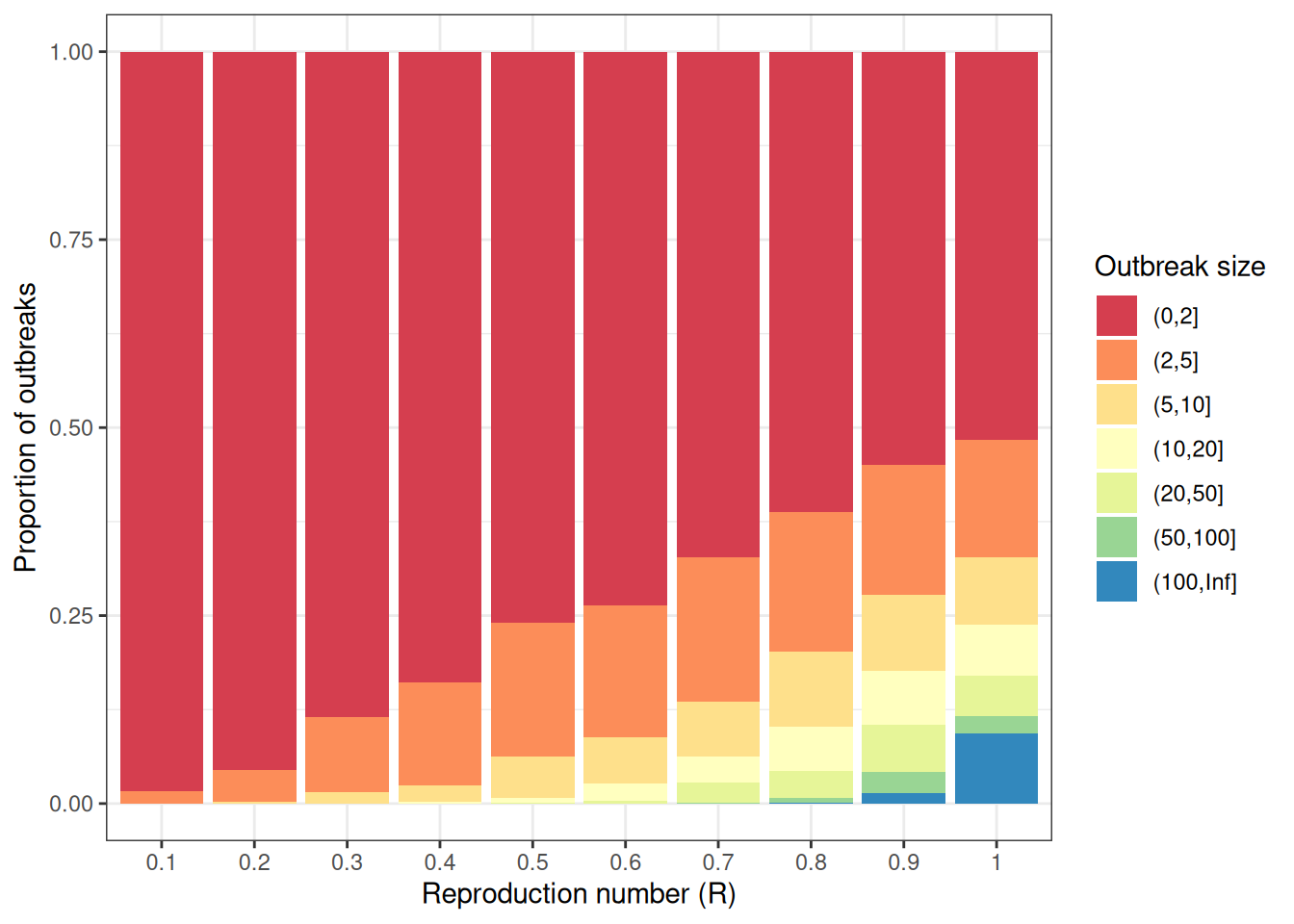
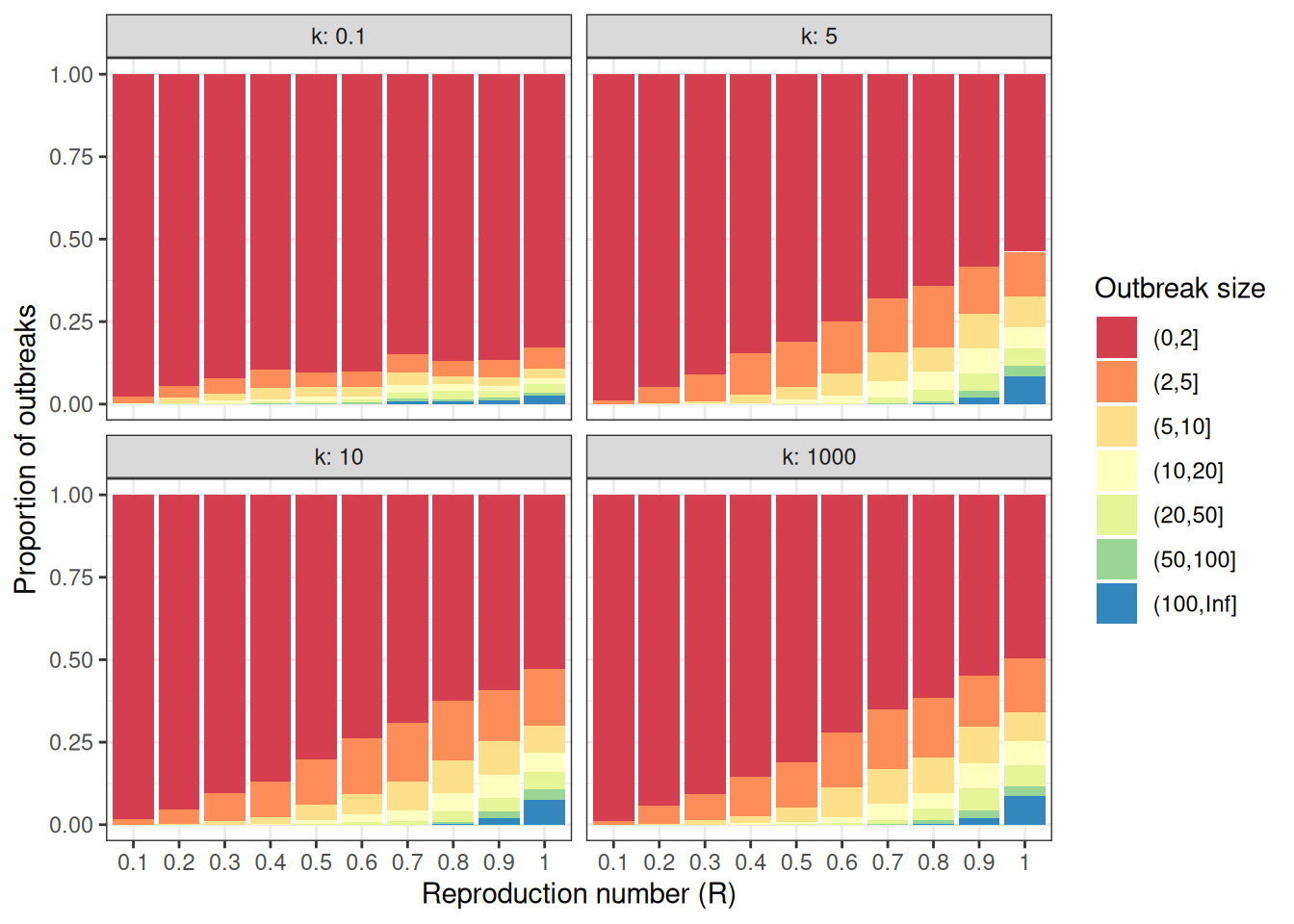
# Change offspring distribution to Negative binomial ----------------------statistic<-"size"offspring_dist<-"rnbinom"R<-seq(0.1, 1.0, 0.1)k<-c(0.1, 5, 10, 1000)scenarios<-expand.grid( offspring_dist =offspring_dist, statistic =statistic, R =R, k =k, stringsAsFactors =FALSE)# Simulate outbreak size distribution (Negative binomial) -----------------outbreak_list<-vector(mode ="list", length =nrow(scenarios))for(iinseq_len(nrow(scenarios))){offspring_dist_fun<-match.fun(scenarios[i, "offspring_dist"])outbreak_list[[i]]<-epichains::simulate_chain_stats( n_chains =n_chains, statistic =scenarios[i, "statistic"], offspring_dist =offspring_dist_fun, mu =scenarios[i, "R"], size =scenarios[i, "k"], stat_threshold =breaks[length(breaks)-1]+1)}# Group outbreak sizes ----------------------------------------------------intervals<-lapply(outbreak_list, cut, breaks =breaks)prop<-lapply(intervals, function(interval){table(interval)/sum(table(interval))})outbreak_size_list<-lapply(prop, as.data.frame)for(iinseq_len(nrow(scenarios))){outbreak_size_list[[i]]$R<-scenarios[i, "R"]outbreak_size_list[[i]]$k<-scenarios[i, "k"]outbreak_size_list[[i]]$offspring_dist<-scenarios[i, "offspring_dist"]outbreak_size_list[[i]]$statistic<-scenarios[i, "statistic"]}outbreak_size2<-do.call(rbind, outbreak_size_list)head(outbreak_size2)#> interval Freq R k offspring_dist statistic#> 1 (0,2] 0.978 0.1 0.1 rnbinom size#> 2 (2,5] 0.019 0.1 0.1 rnbinom size#> 3 (5,10] 0.002 0.1 0.1 rnbinom size#> 4 (10,20] 0.001 0.1 0.1 rnbinom size#> 5 (20,50] 0.000 0.1 0.1 rnbinom size#> 6 (50,100] 0.000 0.1 0.1 rnbinom size# Plot outbreak size distribution (Negative binomial) ---------------------ggplot2::ggplot(data =outbreak_size2)+ggplot2::geom_col( mapping =ggplot2::aes(x =as.factor(R), y =Freq, fill =interval))+ggplot2::scale_x_discrete(name ="Reproduction number (R)")+ggplot2::scale_y_continuous(name ="Proportion of outbreaks")+ggplot2::scale_fill_brewer( name ="Outbreak size", palette ="Spectral")+ggplot2::facet_wrap( facets =c("k"), labeller =ggplot2::label_both)+ggplot2::theme_bw()
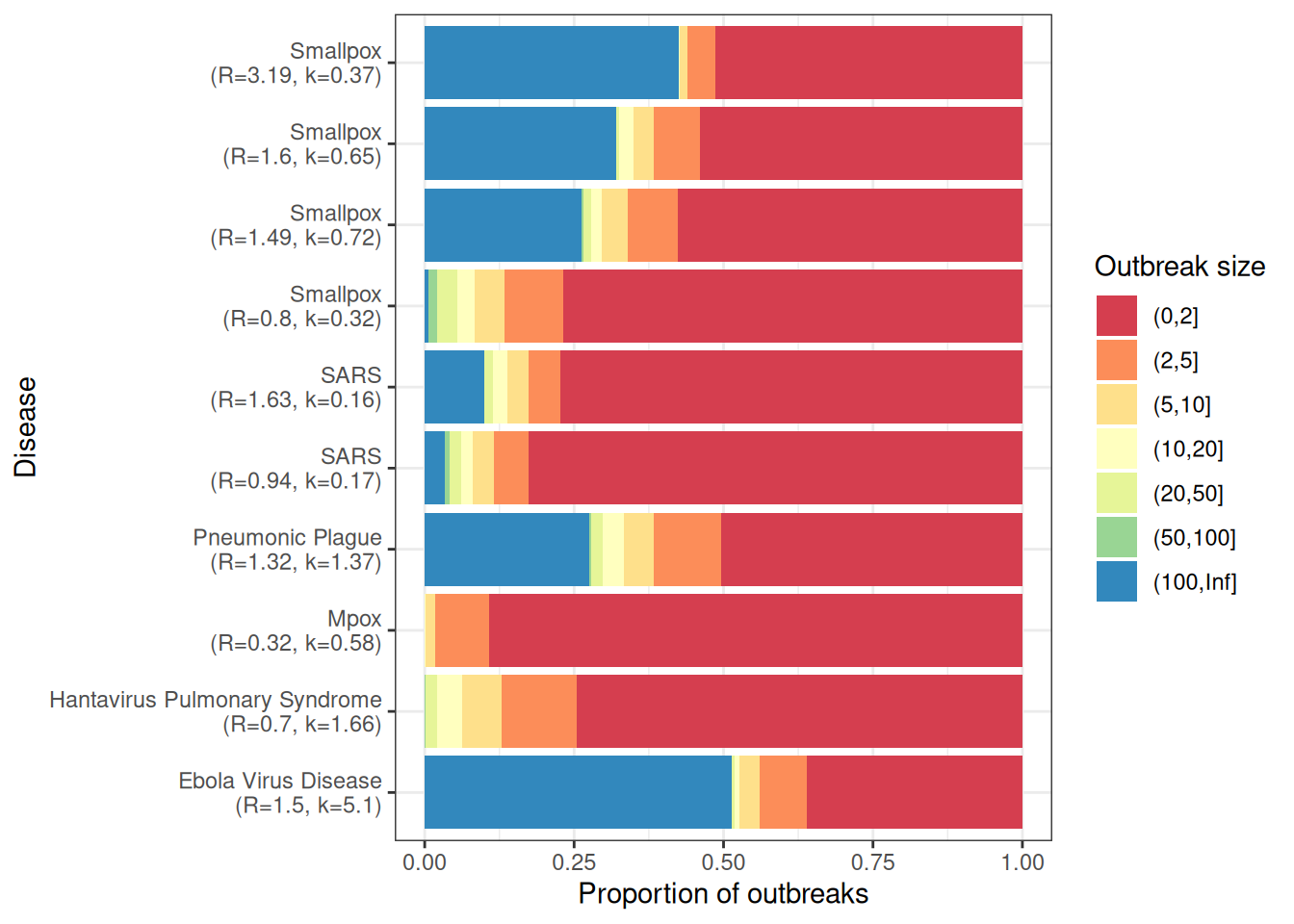
# Simulating outbreak sizes for past outbreaks ----------------------------offspring_dists<-epiparameter::epiparameter_db( epi_name ="offspring distribution")# Check all empirical distributions are the same --------------------------length(unique(vapply(offspring_dists, family, FUN.VALUE =character(1))))==1#> [1] TRUE# Simulate empirical outbreak size distributions --------------------------outbreak_list<-vector(mode ="list", length =length(offspring_dists))for(iinseq_along(offspring_dists)){offspring_dist_fun<-match.fun(paste0("r", family(offspring_dists[[i]])))outbreak_list[[i]]<-epichains::simulate_chain_stats( n_chains =n_chains, statistic ="size", offspring_dist =offspring_dist_fun, mu =epiparameter::get_parameters(offspring_dists[[i]])[["mean"]], size =epiparameter::get_parameters(offspring_dists[[i]])[["dispersion"]], stat_threshold =breaks[length(breaks)-1]+1)}# Group outbreak sizes ----------------------------------------------------# paste suffix as some diseases have multiple offspring distributionsdiseases<-make.unique(vapply(offspring_dists, `[[`, FUN.VALUE =character(1), "disease"))intervals<-lapply(outbreak_list, cut, breaks =breaks)prop<-lapply(intervals, function(interval){table(interval)/sum(table(interval))})outbreak_size_list<-lapply(prop, as.data.frame)for(iinseq_along(offspring_dists)){outbreak_size_list[[i]]$R<-epiparameter::get_parameters(offspring_dists[[i]])[["mean"]]outbreak_size_list[[i]]$k<-epiparameter::get_parameters(offspring_dists[[i]])[["dispersion"]]outbreak_size_list[[i]]$offspring_dist<-family(offspring_dists[[i]])outbreak_size_list[[i]]$disease<-diseases[i]outbreak_size_list[[i]]$statistic<-"size"}outbreak_size3<-do.call(rbind, outbreak_size_list)outbreak_size3<-outbreak_size3|>dplyr::mutate( disease =stringr::str_remove(disease, "\\.\\d+$"), interval =factor(interval, levels =unique(interval)), # preserve order# Create a label combining R and k for each disease disease_label =paste0(disease, "\n(R=", R, ", k=", k, ")"))head(outbreak_size3)#> interval Freq R k offspring_dist disease statistic#> 1 (0,2] 0.773 1.63 0.16 nbinom SARS size#> 2 (2,5] 0.054 1.63 0.16 nbinom SARS size#> 3 (5,10] 0.034 1.63 0.16 nbinom SARS size#> 4 (10,20] 0.024 1.63 0.16 nbinom SARS size#> 5 (20,50] 0.015 1.63 0.16 nbinom SARS size#> 6 (50,100] 0.001 1.63 0.16 nbinom SARS size#> disease_label#> 1 SARS\n(R=1.63, k=0.16)#> 2 SARS\n(R=1.63, k=0.16)#> 3 SARS\n(R=1.63, k=0.16)#> 4 SARS\n(R=1.63, k=0.16)#> 5 SARS\n(R=1.63, k=0.16)#> 6 SARS\n(R=1.63, k=0.16)# Plot empirical outbreak sizes -------------------------------------------ggplot2::ggplot(data =outbreak_size3)+ggplot2::geom_col( mapping =ggplot2::aes( x =as.factor(disease_label), y =Freq, fill =interval))+ggplot2::scale_x_discrete( name ="Disease")+ggplot2::scale_y_continuous(name ="Proportion of outbreaks")+ggplot2::scale_fill_brewer( name ="Outbreak size", palette ="Spectral")+coord_flip()+ggplot2::theme_bw()
Steps in detail
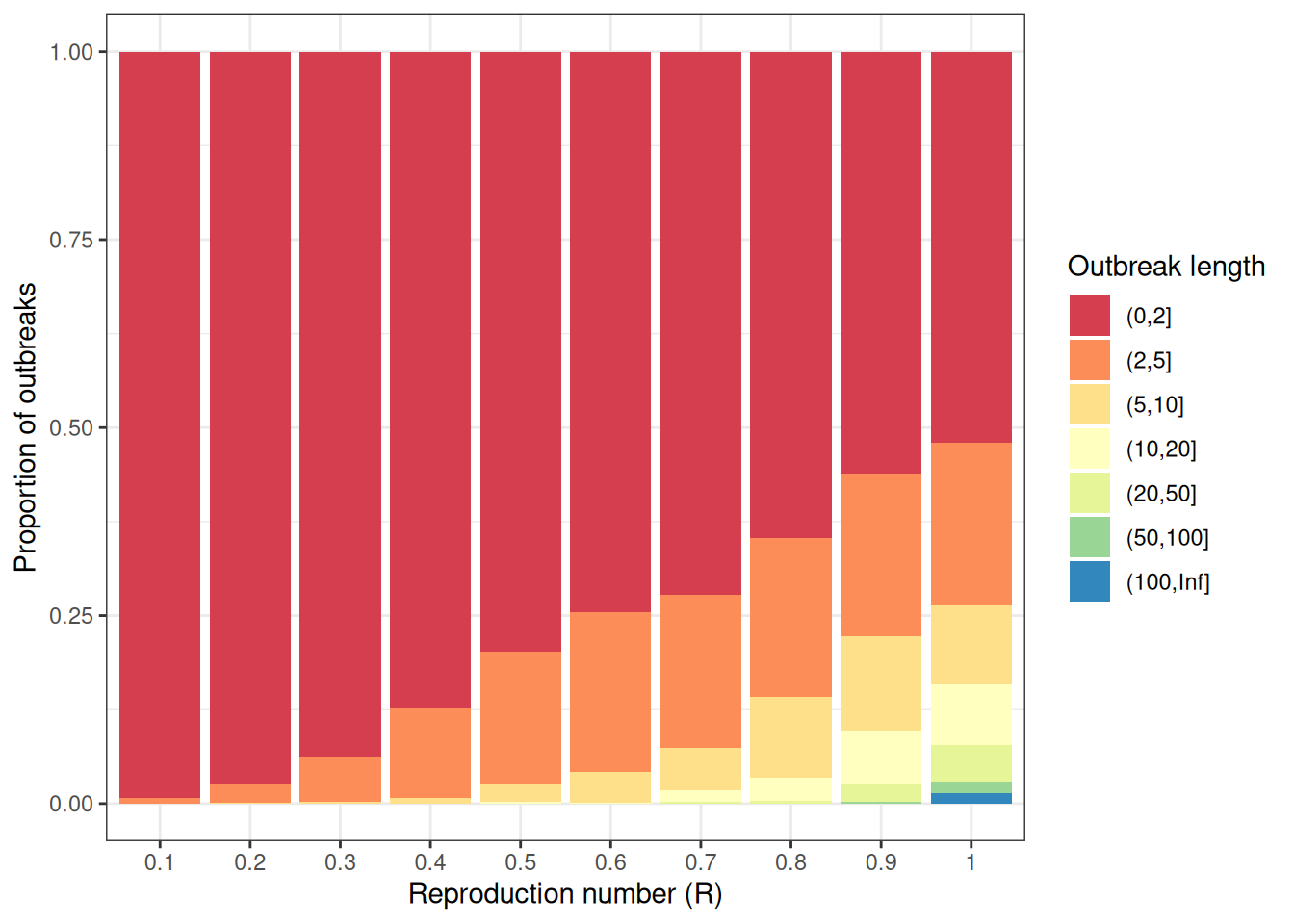
Size is defined as the total number of individuals infected across all generations of infection.
Please note that the code assumes the necessary packages are already installed. If they are not, you can install them using first the install.packages("pak") function and then the pak::pak() function for both packages in CRAN or GitHub before loading them with library().