Projecting infectious disease incidence: a COVID-19 example
James Azam, Sebastian Funk
Source:vignettes/projecting_incidence.Rmd
projecting_incidence.RmdOverview
Branching processes can be used to project stochastic infectious
disease trends in time provided we can characterize the distribution of
times between successive cases (serial interval), and the distribution
of secondary cases produced by a single individual (offspring
distribution). Such simulations can be achieved in epichains
with the simulate_chains() function and Pearson et al. (2020), and Abbott et al. (2020) illustrate its application to
COVID-19.
The purpose of this vignette is to use early data on COVID-19 in South Africa (Marivate and Combrink 2020) to illustrate how epichains can be used to project an outbreak.
Let’s load the required packages
Data
Included in epichains is a cleaned time series of the first 15 days of the COVID-19 outbreak in South Africa. This can be loaded into memory as follows:
data("covid19_sa", package = "epichains")We will use the first \(5\) observations for this demonstration. We will assume that all the cases in that subset are imported and did not infect each other.
Let us subset and view that aspect of the data.
seed_cases <- covid19_sa[1:5, ]
head(seed_cases)
#> # A tibble: 5 × 2
#> date cases
#> <date> <int>
#> 1 2020-03-05 1
#> 2 2020-03-07 1
#> 3 2020-03-08 1
#> 4 2020-03-09 4
#> 5 2020-03-11 6Setting up the inputs
We will now proceed to set up simulate_chains() for the
simulations.
Onset times
simulate_chains() requires a vector of seeding times,
t0, for each chain/individual/simulation.
To get this, we will use the observation date of the index case as the reference and find the difference between the other observed dates and the reference.
days_since_index <- as.integer(seed_cases$date - min(seed_cases$date))
days_since_index
#> [1] 0 2 3 4 6Using the vector of start times from the time series, we will then
create a corresponding seeding time for each individual, which we’ll
call t0.
t0 <- rep(days_since_index, seed_cases$cases)
t0
#> [1] 0 2 3 4 4 4 4 6 6 6 6 6 6Generation time
In epidemiology, the generation time (also called the generation interval) is the duration between successive infectious events in a chain of transmission. Similarly, the serial interval is the duration between observed symptom onset times between successive cases in a transmission chain. The generation interval is often hard to observe because exact times of infection are hard to measure hence, the serial interval is often used instead. Here, we use the serial interval and interpret the simulated case data to represent symptom onset.
In this example, we will assume based on COVID-19 literature that the serial interval, S, is log-normal distributed with parameters, \(\mu = 4.7\) and \(\sigma = 2.9\) (Pearson et al. 2020). The log-normal distribution is commonly used in epidemiology to characterise quantities such as the serial interval because it has a large variance and can only be positive-valued (Nishiura 2007; Limpert et al. 2001).
Note that when the distribution is described this way, it means \(\mu\) and \(\sigma\) are the expected value and standard deviation of the natural logarithm of the serial interval. Hence, in order to sample the “back-transformed” measured serial interval with expectation/mean, \(E[S]\) and standard deviation, \(SD [S]\), we can use the following parametrisation:
\[\begin{align} E[S] &= \ln \left( \dfrac{\mu^2}{(\sqrt{\mu^2 + \sigma^2}} \right) \\ SD [S] &= \sqrt {\ln \left(1 + \dfrac{\sigma^2}{\mu^2} \right)} \end{align}\]
See “log-normal_distribution” on Wikipedia for a detailed explanation of this parametrisation.
We will now set up the generation time function with the appropriate
inputs. We adopt R’s random lognormal distribution generator
(rlnorm()) that takes meanlog and
sdlog as arguments, which we define with the
parametrisation above as log_mean() and
log_sd() respectively and wrap it in the
generation_time_fn() function. Moreover,
generation_time_fn() takes one argument n as
is required by epichains (See
?epichains::simulate_chains), which is further passed to
rlnorm() as the first argument to determine the number of
observations to sample (See ?rlnorm).
Offspring distribution
Let us now set up the offspring distribution, that is the distribution that drives the mechanism behind how individual cases infect other cases. The appropriate way to model the offspring distribution is to capture both the population-level transmissibility (\(R0\)) and the individual-level heterogeneity in transmission (“superspreading”). The negative binomial distribution is commonly used in this case (Lloyd-Smith et al. 2005).
For this example, we will assume that the offspring distribution is characterised by a negative binomial with \(mu = 2.5\) (Abbott et al. 2020) and \(size = 0.58\) (Wang et al. 2020).
mu <- 2.5
size <- 0.58In this parameterization, \(mu\) represents the \(R_0\), which is defined as the average number of cases produced by a single individual in an entirely susceptible population. The parameter \(size\) represents superspreading, that is, the degree of heterogeneity in transmission by single individuals.
Simulation controls
For this example, we will simulate outbreaks that end \(21\) days after the last date of
observations in the seed_cases dataset.
#' Date to end simulation
projection_window <- 21
tf <- max(days_since_index) + projection_window
tf
#> [1] 27simulate_chains() is stochastic, meaning the results are
different every time it is run for the same set of parameters. We will,
therefore, run the simulations \(100\)
times and aggregate the results.
Let us specify that.
#' Number of simulations
sim_rep <- 100Lastly, since, we have specified that \(R0 > 1\), it means the epidemic could potentially grow without end. Hence, we must specify an end point for the simulations.
simulate_chains() provides the
stat_threshold argument for this purpose. Above
stat_threshold, the simulation is cut off. If this value is
not specified, it assumes a value of infinity. Here, we will assume a
maximum chain size of \(1000\).
#' Maximum chain size allowed
stat_threshold <- 1000Modelling assumptions
This exercise makes the following simplifying assumptions:
- All cases are observed.
- Cases are observed exactly at the time of infection.
- There is no reporting delay.
- Reporting rate is constant through the course of the epidemic.
- No interventions have been implemented.
- Population is homogeneous and well-mixed.
To summarise the whole set up so far, we are going to simulate each chain 100 times, projecting cases over 21 days after the first 6 days, and assuming that no outbreak size exceeds 1000 cases.
Running the simulations
We will use the function lapply() to run the simulations
and bind them by rows with dplyr::bind_rows().
set.seed(1234)
sim_chain_sizes <- lapply(
seq_len(sim_rep),
function(sim) {
simulate_chains(
n_chains = length(t0),
offspring_dist = rnbinom,
mu = mu,
size = size,
statistic = "size",
stat_threshold = stat_threshold,
generation_time = generation_time,
t0 = t0,
tf = tf
) %>%
mutate(sim = sim)
}
)
sim_output <- bind_rows(sim_chain_sizes)Let us view the first few rows of the simulation results.
head(sim_output)
#> chain infector infectee generation time sim
#> 14 3 1 2 2 8.123133 1
#> 15 3 1 3 2 6.842933 1
#> 16 3 1 4 2 7.260639 1
#> 17 3 1 5 2 11.244838 1
#> 18 7 1 2 2 7.022334 1
#> 19 7 1 3 2 12.182368 1Post-processing
Now, we will summarise the simulation results.
We want to plot the individual simulated daily time series and show the median cases per day aggregated over all simulations.
First, we will create the daily time series per simulation by aggregating the number of cases per day of each simulation.
# Daily number of cases for each simulation
incidence_ts <- sim_output %>%
mutate(day = ceiling(time)) %>%
count(sim, day, name = "cases") %>%
as_tibble()
head(incidence_ts)
#> # A tibble: 6 × 3
#> sim day cases
#> <int> <dbl> <int>
#> 1 1 0 1
#> 2 1 2 1
#> 3 1 3 1
#> 4 1 4 4
#> 5 1 6 6
#> 6 1 7 1Next, we will add a date column to the results of each simulation set. We will use the date of the first case in the observed data as the reference start date.
# Get start date from the observed data
index_date <- min(seed_cases$date)
index_date
#> [1] "2020-03-05"
# Add a dates column to each simulation result
incidence_ts_by_date <- incidence_ts %>%
group_by(sim) %>%
mutate(date = index_date + days(seq(0, n() - 1))) %>%
ungroup()
head(incidence_ts_by_date)
#> # A tibble: 6 × 4
#> sim day cases date
#> <int> <dbl> <int> <date>
#> 1 1 0 1 2020-03-05
#> 2 1 2 1 2020-03-06
#> 3 1 3 1 2020-03-07
#> 4 1 4 4 2020-03-08
#> 5 1 6 6 2020-03-09
#> 6 1 7 1 2020-03-10Now we will aggregate the simulations by day and evaluate the median daily cases across all simulations.
# Median daily number of cases aggregated across all simulations
median_daily_cases <- incidence_ts_by_date %>%
group_by(date) %>%
summarise(median_cases = median(cases)) %>%
ungroup() %>%
arrange(date)
head(median_daily_cases)
#> # A tibble: 6 × 2
#> date median_cases
#> <date> <dbl>
#> 1 2020-03-05 1
#> 2 2020-03-06 1
#> 3 2020-03-07 1
#> 4 2020-03-08 4
#> 5 2020-03-09 5
#> 6 2020-03-10 8Visualization
We will now plot the individual simulation results alongside the median of the aggregated results.
# since all simulations may end at a different date, we will find the minimum
# final date for all simulations for the purposes of visualisation.
final_date <- incidence_ts_by_date %>%
group_by(sim) %>%
summarise(final_date = max(date), .groups = "drop") %>%
summarise(min_final_date = min(final_date)) %>%
pull(min_final_date)
incidence_ts_by_date <- incidence_ts_by_date %>%
filter(date <= final_date)
median_daily_cases <- median_daily_cases %>%
filter(date <= final_date)
ggplot(data = incidence_ts_by_date) +
geom_line(
aes(
x = date,
y = cases,
group = sim
),
color = "grey",
linewidth = 0.2,
alpha = 0.25
) +
geom_line(
data = median_daily_cases,
aes(
x = date,
y = median_cases
),
color = "tomato3",
linewidth = 1.8
) +
geom_point(
data = covid19_sa,
aes(
x = date,
y = cases
),
color = "black",
size = 1.75,
shape = 21
) +
geom_line(
data = covid19_sa,
aes(
x = date,
y = cases
),
color = "black",
linewidth = 1
) +
scale_x_continuous(
breaks = seq(
min(incidence_ts_by_date$date),
max(incidence_ts_by_date$date),
5
),
labels = seq(
min(incidence_ts_by_date$date),
max(incidence_ts_by_date$date),
5
)
) +
scale_y_continuous(
breaks = seq(
0,
max(incidence_ts_by_date$cases),
30
),
labels = seq(
0,
max(incidence_ts_by_date$cases),
30
)
) +
geom_vline(
mapping = aes(xintercept = max(seed_cases$date)),
linetype = "dashed"
) +
labs(x = "Date", y = "Daily cases")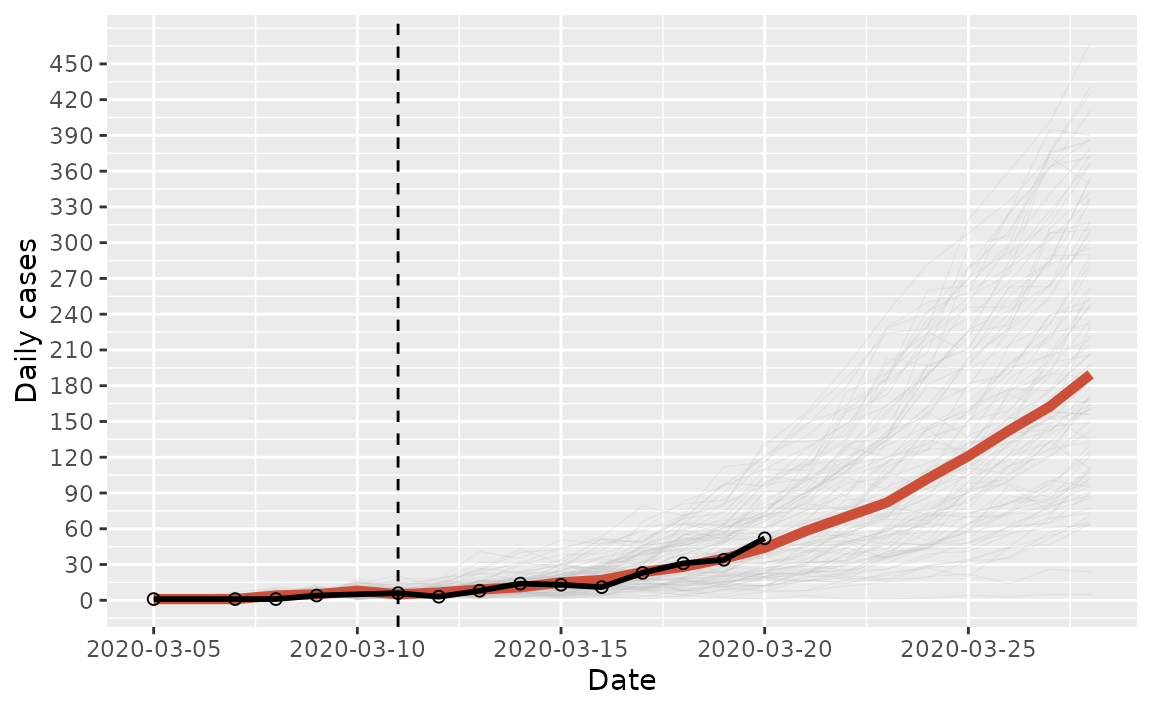
COVID-19 incidence in South Africa projected over a two week window in 2020. The light gray lines represent the individual simulations, the red line represents the median daily cases across all simulations, the black connected dots represent the observed data, and the dashed vertical line marks the beginning of the projection.